Ensure atom names are unique after construction #469
Conversation
…lowing construction; ensure atom names are unique after construction
|
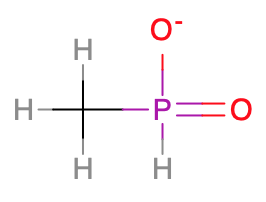
Correction: The minidrugbank phosphorous-containing molecule actually has a stereocenter because its substituents are different: With the RDKit backend: >>> Molecule.from_smiles('[H]C([H])([H])P(=O)([H])[O-]')
---------------------------------------------------------------------------
UndefinedStereochemistryError Traceback (most recent call last)
<ipython-input-14-9e8e428709fa> in <module>
----> 1 Molecule.from_smiles('[H]C([H])([H])P(=O)([H])[O-]')
~/miniconda/lib/python3.7/site-packages/openforcefield/topology/molecule.py in from_smiles(smiles, hydrogens_are_explicit, toolkit_registry, allow_undefined_stereo)
~/miniconda/lib/python3.7/site-packages/openforcefield/utils/toolkits.py in call(self, method_name, *args, **kwargs)
~/miniconda/lib/python3.7/site-packages/openforcefield/utils/toolkits.py in from_smiles(self, smiles, hydrogens_are_explicit, allow_undefined_stereo)
~/miniconda/lib/python3.7/site-packages/openforcefield/utils/toolkits.py in _detect_undefined_stereo(cls, rdmol, err_msg_prefix, raise_warning)
UndefinedStereochemistryError: Unable to make OFFMol from RDMol: Unable to make OFFMol from SMILES: RDMol has unspecified stereochemistry. Undefined chiral centers are:
- Atom P (index 4)
Perhaps we shouldn't be using minidrugbank, or we should filter out the compounds that contain unspecified stereochemistry? |
|
The remaining test failures appear to be due to the SMILES round-trip tests failing because we have some molecules in minidrugbank that have unspecified stereochemistry, yet we load them all in for testing. What approach should we take with these? Should we skip molecules with unspecified stereochemistry in our round-trip tests? |
 j-wags
left a comment
j-wags
left a comment
There was a problem hiding this comment.
Sorry for delay, this somehow fell off my radar last week.
These changes look technically solid. My one major question is whether we want unique naming to always happen, or to be something that the user chooses to apply (they could call offmol.generate_unique_atom_names manually if they need it).
I think that making the user call it manually is more in line with our philosophy, since
- We will in some cases surprise users by overwriting their atom names, since we never told them they need to be unique (and who knows why they named the atoms the way they did)
- Atom names have no functional effect on how we parametrize a system, so enforcing rules/behavior about them is misleading at best
- A user could "invalidate" a molecule by loading it (so it has unqiue atom names), and then setting one atom's name equal to another's. Then the molecule would fail a round-trip test.
I'm worried that our philosophy is trending toward "annoy the hell out of the user by preventing them from ever doing anything easily with lots of small molecules". As it is right now, the user cannot use our tools to, say,
I agree that the philosophy of "raise an informative Exception subclass object for each error type that the user must correct" can be an effective way to make sure the user is informed of each and every criteria their molecules must meet to be properly processed, but I can guarantee that this is going to cause a great deal of frustration. Is there some way we can provide both a "paranoid mode" (where we ensure that we are typing exactly the molecule the user wants) and some sort of "just process these molecules mode" where molecules that don't pass our strict criteria are either transformed into a processable form or are dropped from the list? Even dropping may be problematic, because---as was pointed out previously---we are standardizing on SDF files, which do not have atom names, which I believe makes it impossible to transform the resulting system to AMBER/gromacs with ParmEd. If we were following the "principle of least surprise", I'd probably suggest the behavior where missing atom names are filled in silently with unique atom names that do not collide with any existing atom names, but an exception is raised if two user-specified atom names are identical. That would allow processing of molecules without issue in a manner that would not change any atom names the user specified, but would raise an exception if the user-specified atom names caused a problem. Otherwise, they'd be filled in so things just work. |
|
I noted this above, but it's important enough to state again here: Once we ditch ParmEd, it will no longer be necessary to generate unique atom names. But until we replace it, we're stick with the need to do so if we want our parameters to be deliverable to different molecular simulation packages. That means that this needs to either happen to all molecules we load into the toolkit, or it means we need to create more code to manage the transformation to other packages that does this silently behind the scenes. That may be an alternative to this approach that is workable! |
|
I wrote a lot, but it boils down to this:
I think we should do this, at the time of system creation. So, either we
|
|
To be a little less brief on my reasoning: I think we need to keep our internal data structures clear of ParmEd's influence. The above discussion was driving us to choose a group of users who would "lose". Either:
When really, we can just push this battle outside our borders -- Make the users identify where these parameters are going, and apply naming that works for ParmEd if that's the destination. And maybe the second option doesn't seem too bad, but the slope toward ParmEd-ness becomes no less steep if we accept that behavior. We just get into the "if you give a moose a muffin" situation -- If we rename the alanine atoms above, users will say "well, they're just in different residues, OFFTK should allow that". Then we'll be pushed to enforce some system of rules around atom names and residue names/numbers, in a toolkit made specifically not to care about atom types or topological residues. People will make workflows assuming the atom names we assign, and contort their inputs to satisfy the residue rules we enforce, and then we'll have this large set of expected behaviors causing friction on further developments (do virtual sites also need unique names? do we support chains?). On a separate note: I agree that the undefined stereochemistry thing is ugly and awkward. It may not be worth it on some fronts. I'd be open to arguments that we should just drop it for trivalent nitrogen and sulfur -- We could say stereochemistry that exchanges more frequently than once per millisecond is not stereochemistry at all for the purposes of atomistic simulation. This will be made easier by breaking out a |
|
@j-wags : I like your suggestions in #469 (comment)! I'd be fine with this, but don't have the bandwidth to re-work this PR anytime soon. Should be an easy fix, though! The only thing I'd caution about is that the wider the separation between doing the thing that triggers an error (converting a molecule with defective atom names) and the error (creating a system), the harder it is for our users to identify where the error was actually committed! So we have to help them along as much as possible by being maximally informative in our error messages.
I still like the idea of an Another approach that would make our lives easier is to just say we're not going to care about stereochemistry in charge models, and that any model we ultimately adopt would have to somehow average over all possible stereochemical configurations. That would make the cheminformatics easy, but it may ultimately limit accuracy. |
|
@j-wags: How should we proceed here? We're not going to be able to use SDF files and ParmEd to convert to other packages unless we implement something like this now, and I'd love to get this finished before the |
|
@j-wags: How should we proceed here? |
I'll:
|
|
Oh, OpenMM
|
This PR adds a
Molecule._validate()method that is called after all molecule construction events as a way to run any code required to standardize or otherwise validate the constructedMoleculeobject.This PR also fixes #460 by ensuring that unique atom names are generated at this time if the specified atom names are not unique. Note that this may cause atom names to change if some were defined, but not all were unique.
This is done via addition of
Molecule.has_unique_atom_namesproperty and aMolecule.generate_unique_atom_names()public method.There are some alternative strategies discussed in #460, but as #460 (comment) points out, if we're going to be standardizing on SDF files (which lack atom names), we need to do something to ensure unique atom names are generated by the time we need to use them.
Additionally, two tests were added:
The failing test here also involves a phosphorous containing molecule (
[H]C([H])([H])P(=O)([H])[O-]) suggesting #467 extends to phosphorous as well.