Input Files
Evidente requires two files, a phylogenetic tree (Newick format) and a SNP table (TSV/CSV file that summarises VCF files). Evidente can be used to visualize metadata ( in a TSV/CSV file), and can take an annotation file (GFF file) and GO-terms as an input for an enrichment analysis.
In this section we describe the input files of evidente. The main input of Evidente consists of:
- A phylogenetic tree in a Newick format
- A SNP table
Optional files are:
- A metadata table (CSV/TSV)
- An annotation file (GFF file)
- A GO Annotation file (CSV/TSV)
All excerpts of the files originate from the toy example preloaded in Evidente.
Any phylogenetic tree in a Newick format can be provided as input. It is important that the file describes the distances between the nodes of the phylogenetic tree.
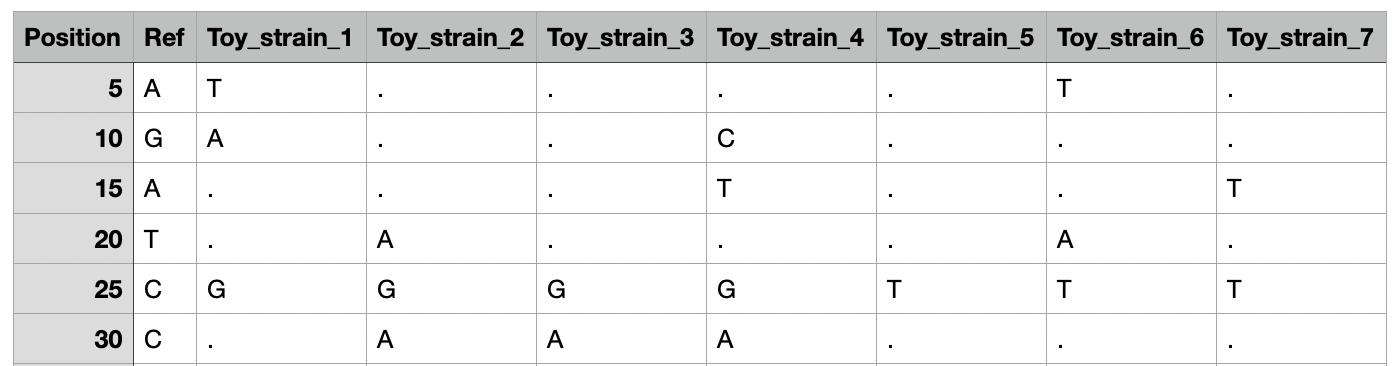
A SNP table is a summary of multiple VCF files as a CSV/TSV format. Such a table can be produced using MUSIAL, but any other tool that delivers the correct output would work. The first column of a SNP table indicates the position of a SNP. The second column (REF) indicates the allele of the reference genome at the given position. All other columns have as a header the name of the samples and indicate the presence of a SNP at the given position. Any base (A/C/T/G) indicates a presence of a SNP, while a dot (".") states that the sample contains the reference allele at that position. Evidente is also able to visualize unresolved bases (N).
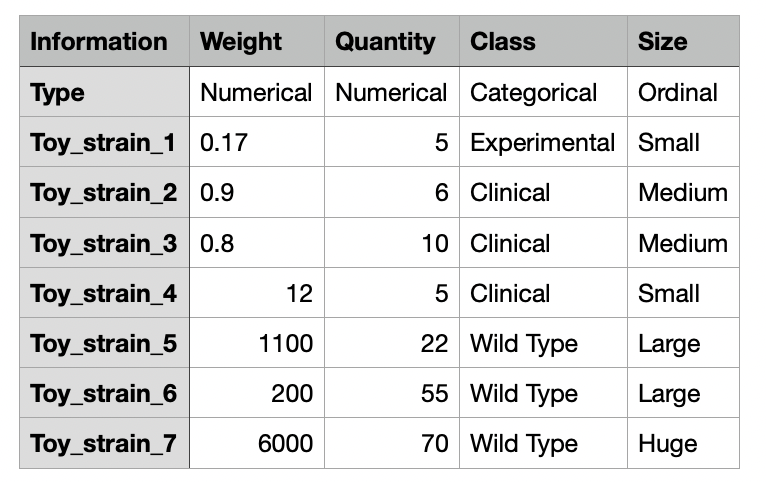
The metadata table is an optional input CSV or TSV file for Evidente. For a correct parsing of the data, the metadata table should be structured as following. The first row should start with a title "Information" and followed by the name of the metadata available. The second column should start with the title "Type" and should contain for each metadata the corresponding category of data. For this classification, we have decided to use the data types presented by Munzner (2014). Hence each metadata should be categorized into Categorical, Ordinal or Numerical.
An annotation file that includes the "locus tag" for each gene in the 9th column. This locus tag is then used for the linkage of the GO-data.

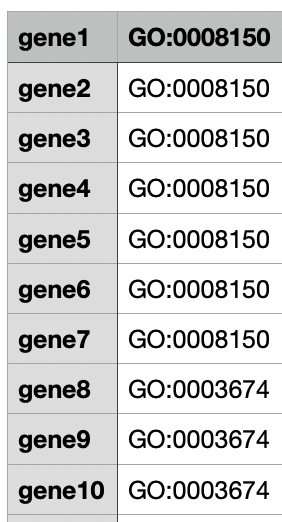
A simple tabular file, where the first column corresponds to the locus tag of a gene, and the second column corresponds to one of the corresponding GO-term. If a gene is linked to many GO-terms, many entries can be created for each gene/locus_tag.