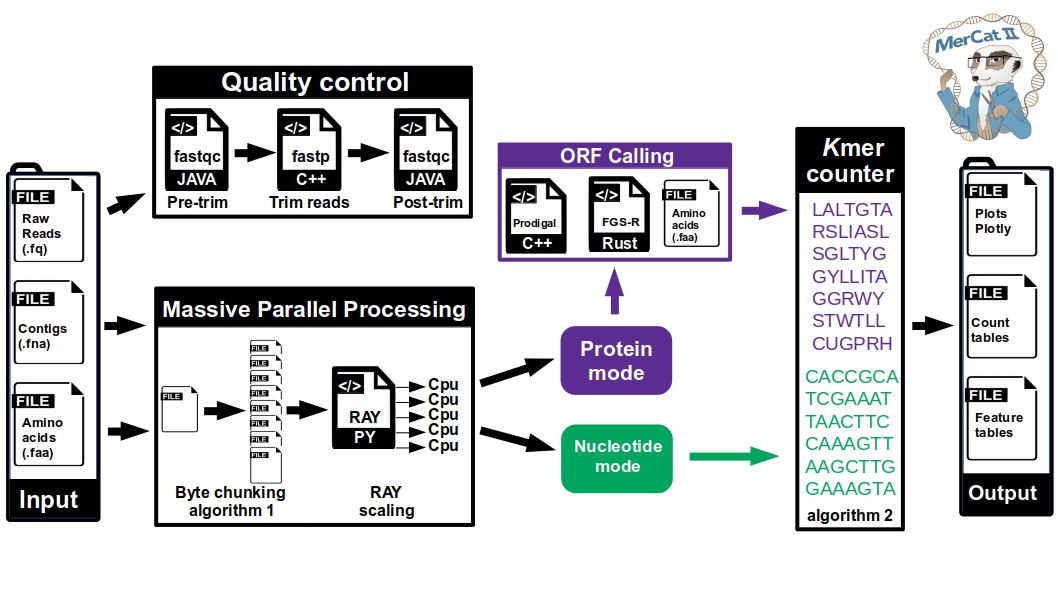
MerCat2: python code for versatile k-mer counter for database independent property analysis (DIPA) for omic analysis






conda activate base
conda install mamba- NOTE: Make sure you install mamba in your base conda environment
- We have found that mamba is faster than conda for installing packages and creating environments. Using conda might fail to resolve dependencies.
- Available via Bioconda:
mamba create -n mercat2 -c conda-forge -c bioconda mercat2
conda activate mercat2- Clone mercat2 from github
- Run install_mercat2.sh to install all required dependencies
- This script creates a conda environment for you
git clone https://github.com/raw-lab/mercat2.git
cd mercat2
bash install_mercat2.sh
conda activate mercat2MerCat2 runs on python version 3.9 and higher.
MerCat2 can run without external dependencies based on the options used.
Required dependencies:
-
When a raw read .fastq file is given
-
For bacteria/archaea rich samples (-prod option)
-
For eukaryote rich samples or general applications (-fgs option)
These are available through BioConda, except FragGeneScanRS, which is included in the MerCat2 distribution.
conda install -c bioconda fastqc fastp prodigalusage: mercat2.py [-h] [-i I [I ...]] [-f F] -k K [-n N] [-c C] [-prod] [-fgs] [-s S] [-o O] [-replace] [-lowmem LOWMEM] [-skipclean] [-toupper] [-pca] [--version]
options:
-h, --help show this help message and exit
-i I path to input file(s)
-f F path to folder containing input files
-k K kmer length
-n N no of cores [auto detect]
-c C minimum kmer count [10]
-prod run Prodigal on fasta files
-fgs run FragGeneScanRS on fasta files
-s S Split into x MB files. [100]
-o O Output folder, default = 'mercat_results' in current directory
-replace Replace existing output directory [False]
-lowmem LOWMEM Flag to use incremental PCA when low memory is available. [auto]
-skipclean skip trimming of fastq files
-toupper convert all input sequences to uppercase
-pca create interactive PCA plot of the samples (minimum of 4 fasta files required)
--version, -v show the version number and exitMercat assumes the input file format based on the extension provided
-
raw fastq file: ['.fastq', '.fq']
-
nucleotide fasta: ['.fa', '.fna', '.ffn', '.fasta']
-
amino acid fasta: ['.faa']
-
It also accepts gzipped versions of these filetypes with the added '.gz' suffix
mercat2.py -i file-name.faa -k 3 -c 10mercat2.py -i file-name.fna -k 3 -n 8 -c 10mercat2.py -i file-name.fastq -k 3 -n 8 -c 10mercat2.py -f /path/to/input-folder -k 3 -n 8 -c 10Run on sample with prodigal/FragGeneScanRS option (raw reads or nucleotide contigs - '.fa', '.fna', '.ffn', '.fasta', '.fastq')
mercat2.py -i /path/to/input-file -k 3 -n 8 -c 10 -prodmercat2.py -i /path/to/input-file -k 3 -n 8 -c 10 -fgs- the prodigal and FragGeneScanRS options run the k-mer counter on both contigs and produced amino acids
- Results are stored in the output folder (default 'mercat_results' of the current working directory)
- the 'report' folder contains an html report with interactive plotly figures
- If at least 4 samples are provided a PCA plot will be included in the html report
- the 'tsv' folder contains counts tables in tab separated format
- if protein files are given, or the -prod option, a .tsv file is created for each sample containing k-mer count, pI, Molecular Weight, and Hydrophobicity metrics
- if nucleotide files are given a .tsv file is created for each sample containing k-mer count and GC content
- if .fastq raw reads files are used, a 'clean' folder is created with the clean fasta file.
- if the -prod option is used, a 'prodigal' folder is created with the amino acid .faa and .gff files
- if the -fgs option is used, a 'fgs' folder is created with the amino acid .faa file
- the 'report' folder contains an html report with interactive plotly figures

Alpha and Beta diversity metrics provided by MerCat2 are experimental. We are currently working on the robustness of these measures.
Alpha diversity metrics provided:
- shannon
- simpson
- simpson_e
- goods_coverage
- fisher_alpha
- dominance
- chao1
- chao1_ci
- ace
Beta diversity metrics provided:
- euclidean
- cityblock
- braycurtis
- canberra
- chebyshev
- correlation
- cosine
- dice
- hamming
- jaccard
- mahalanobis
- manhattan (same as City Block in this case)
- matching
- minkowski
- rogerstanimoto
- russellrao
- seuclidean
- sokalmichener
- sokalsneath
- sqeuclidean
- yule
MerCat2 uses a substantial amount of memory when the k-mer is high.
Running MerCat2 on a personal computer using a k-mer length of ~4 should be OK. Total memory usage can be reduced using the Chunker feature (-s option), but keep in mind that in testing when the chunk size is too small (1MB) some of the least significant k-mers will get lost. This does not seem to affect the overall results, but it is something to keep in mind. Using the chunker and reducing the number of CPUs available (-n option) can help reduce memory requirements.
The speed of MerCat2 can be increased when more memory or computer nodes are available on a cluster and using a chunk size of about 100Mb.
This is copyrighted by University of North Carolina at Charlotte, Jose L. Figueroa III, Andrew Redinbo, and Richard Allen White III. All rights reserved. DeGenPrime is a bioinformatic tool that can be distributed freely for academic use only. Please contact us for commerical use. The software is provided “as is” and the copyright owners or contributors are not liable for any direct, indirect, incidental, special, or consequential damages including but not limited to, procurement of goods or services, loss of use, data or profits arising in any way out of the use of this software.
If you are publishing results obtained using MerCat2, please cite:
Figueroa JL*, Redinbo A*, Panyala A, Colby S, Friesen M, Tiemann L, White III RA. 2024.
MerCat2: a versatile k-mer counter and diversity estimator for database-independent property analysis obtained from omics data
Bioinformatics Advances, vbae061 Bioinformatics Advances
*Co-first authors
BioRxiv pre-print
Figueroa JL, Panyala A, Colby S, Friesen M, Tiemann L, White III RA. 2022.
MerCat2: a versatile k-mer counter and diversity estimator for database-independent property analysis obtained from omics data.
bioRxiv
Please send all queries to Jose Luis Figueroa III
Dr. Richard Allen White III
Andrew Redinbo
Or open an issue