To provide bed files for custom gene-level annotation with VEP. This custom script processes a list of ids (HGNC, transcript) or coordinates with associated annotation, into a comprehensive bed file for the corresponding refseq transcripts for each ID entry.
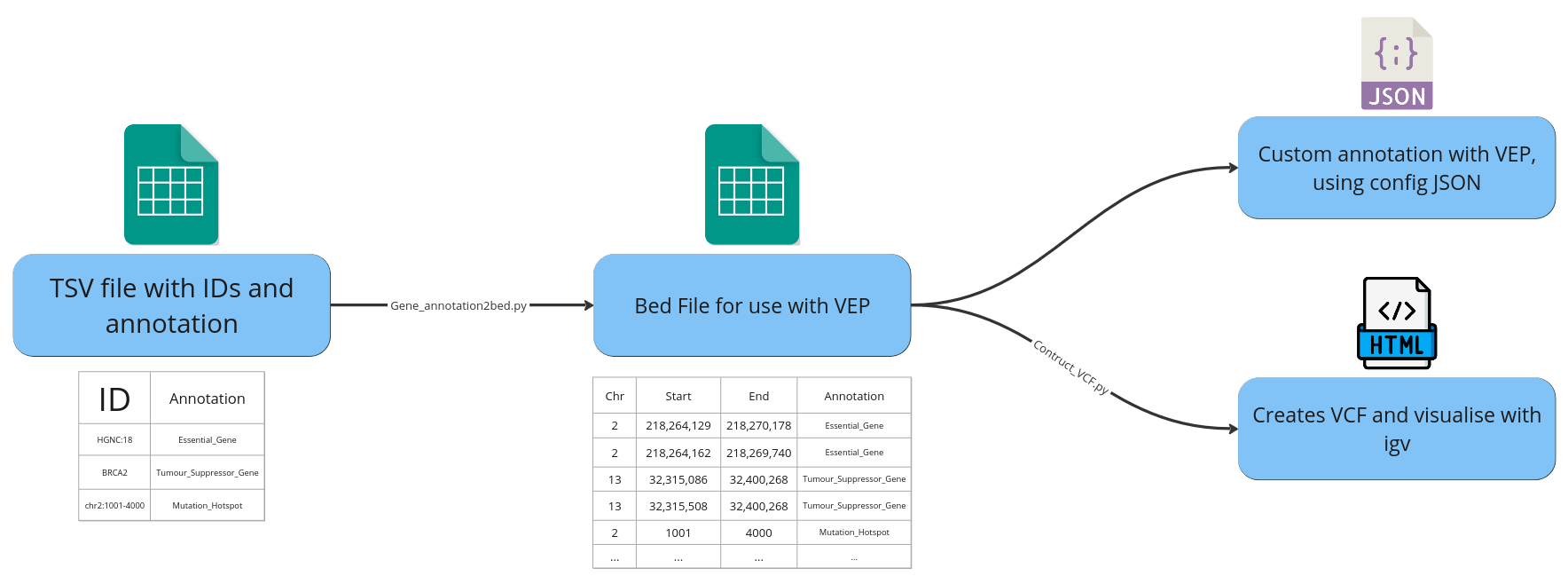
- Converting a list of HGNC ids + associated gene level annotation information into a comprehensive bed file for annotation with Ensemble's VEP.
- Other use cases include providing different inputs such a list of transcripts. Or using exact coordinates to flag a regions such as TERT promoter.
- List of ids and annotation information in TSV format.
- Human Genome Reference (i.e. hs37d5).
- RefSeq Transcripts file (gff3) from 001_reference.
-ig,--annotation_file(str): Path to the annotation file (TSV), this file is essential for the app to execute successfully.-o,output(str): Output file suffix, required for specifying the suffix for the generated output files.-build,--genome_build(str): Reference genome build (hg19/hg38), choose either 'hg19' or 'hg38' based on your requirements.-f,--flanking(int): Flanking size, an integer value representing the size of flanking regions for each gene, transcript or coordinates provided.--assembly_summary(str): Path to assembly summary file, necessary for the app to gather assembly information.-gff(str): Path to GFF file containing all relevant transcripts for assay, available in 001_reference i.e. GCF_000001405.25_GRCh37.p13_genomic.gff.
-ref_igv,--reference_file_for_igv(file): Path to the Reference genome fasta file for igv_reports, used in generating IGV reports.-dump,--hgnc_dump_path(file): Path to HGNC TSV file with HGNC information. Required if gene symbols are present (-gsis specified).
-gs,-symbols_present(bool): Flag to indicate whether gene symbols are present in the annotation file.
-pickle(str): Import GFF as a pickle file, this is for testing mostly to speed-up running, so gff isn't processed each time.
python gene_annotation2bed.py -ig annotation.tsv -o output_suffix -ref hg38 -f 50 --assembly_summary assembly_summary.txt -ref_igv ref_genome.fasta -symbols_present --hgnc_dump_path hgnc_info.tsv -gff your_file.gff -pickle pickle_file.pkl- pysam
- pandas
- igv-reports (v)
- numpy
- re
install using requirements.txt. pip install requirements.txt
IGV report:

The script produces a HTML report of all the bed file entries. Displayed in IGV with the refseq track and bed file aligned with the respecive annotation.
├── data │ ├── demo │ │ ├── after_table.tsv.gz │ │ ├── before_table.tsv.gz │ │ ├── demo_igv_reports.png │ │ └── initial_table.tsv.gz │ ├── GCF_000001405.25_GRCh37.p13_assembly_report.txt │ ├── GCF_000001405.25_GRCh37.p13_genomic.gff │ ├── hg19 │ │ ├── ncbiRefSeq.txt.gz │ │ ├── ncbiRefSeq.txt.gz.tbi │ │ ├── refGene.txt.gz │ │ └── refGene.txt.gz.tbi │ └── hg38 │ ├── ncbiRefSeq.txt.gz │ ├── ncbiRefSeq.txt.gz.tbi │ ├── refGene.txt.gz │ └── refGene.txt.gz.tbi ├── gene_annotation2bed.py (MAIN SCRIPT) ├── LICENSE ├── output_new_test.vcf ├── README.md ├── requirements.txt ├── scripts │ ├── construct_vcf.py │ └── igv_report.py │ ├── tests │ ├── init.py │ ├── test_construct_vcf.py │ ├── test_data │ │ ├── coordinates_anno_test.tsv │ │ ├── example_bed_hg38.bed │ │ ├── expected_output.vcf │ │ ├── hgcn_ids_anno_test.tsv │ │ ├── hs37d5.fa │ │ ├── hs37d5.fa.fai │ │ ├── refseq_gff_preprocessed.pkl │ │ ├── test_empty_attributes.gff │ │ ├── test_empty.gff │ │ ├── test_missing_attributes.gff │ │ └── transcripts_anno_test.tsv │ ├── test_gene_annotation2bed.py │ ├── test_gff_parsing.py │ └── test_igv_report.py │ ├── utils │ ├── configure_gff.py │ ├── gff2pandas.py │ ├── init.py └── Workflow.png