fix #301 #328
fix #301 #328
Conversation
|
Hi @antgonza : the timestamp change has just been merged into master :) so could you please integrate that into this PR and address any check failures that remain afterwards? Would love to review and merge this by the end of the week! |
|
Wait on review, need to do another pass ... |
|
Just restarted the build cause locally the error is random, not sure if related to other issues but it seems like the plate number changes from 2-4 in some occasions, @AmandaBirmingham any ideas? |
|
Yep, I strongly suspect I know the answer to this one. Having looked through the code, I believe that the However, that is not the only issue with this sql statement :), because I see that the known-good prep file does not order samples by sample id at all but rather by plate and then by position (first by column, then by row--the opposite of what the above sql does):
Since the goal is to reproduce the known-good file, |
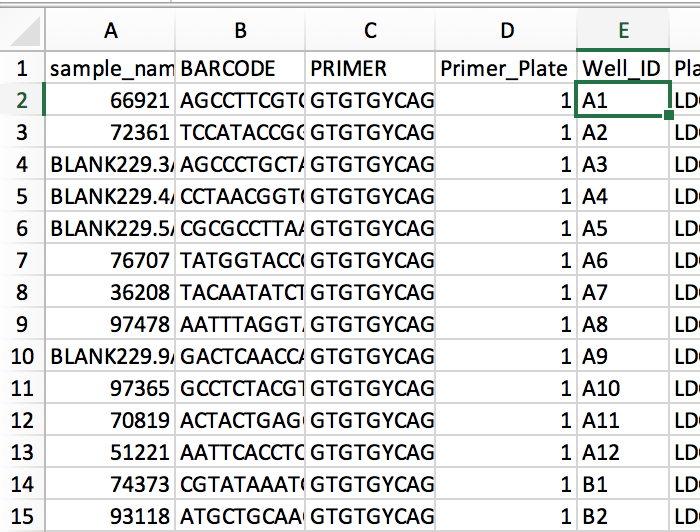
|
Thank and sounds good, any suggestions on how to adequately constrain the SQL? |
|
That said, if you decide you want to open a new issue for the plate/column/row sorting instead of handling it in this PR, I'm ok with that. In that case, it would still be necessary to add an additional constraint to the sql ORDER BY statement, or else to explicitly sort the results in python afterwards, so that the output order becomes deterministic and is thus stable every time we run the tests :) |
|
Ooops, sorry, our messages crossed ... |
|
My initial guess would be that ordering by plate, then col_num, then row_num would do it. I don't know if there is actually supposed to be a study ordering ... the known-good file I have all comes from a single study, so I'd have to check that with the wet lab. |
|
Adding in the plate to ordering should do it, assuming the plate number is globally unique (to labman). Organizing the output by study could be nice for subsequent debugging and human eyes. But, I can also see the value in having it ordered by physical location too. |
|
I think we will want to use the plate external_id (the user-assigned plate id), which I think is what @antgonza already has in
I've reached out to Gail and the wet lab about whether they want additional ordering by study, but until/unless they say they do, I say we stick with straight-up positional ordering for now. |
|
For consistency, should we do sample_plate, primer_plate, col_num, row_num or do you have any other suggestions? |
|
Note that the sorting in SQL is ignored once everything moves to a dataframe, so decided to remove that sorting and use the pandas sorting ... |
|
Ok, so the latest test failure says:
I began debugging by simply pulling your test's
For example, it looks like:
Given this, I think the best place to start fixing this test failure is probably returning to the |
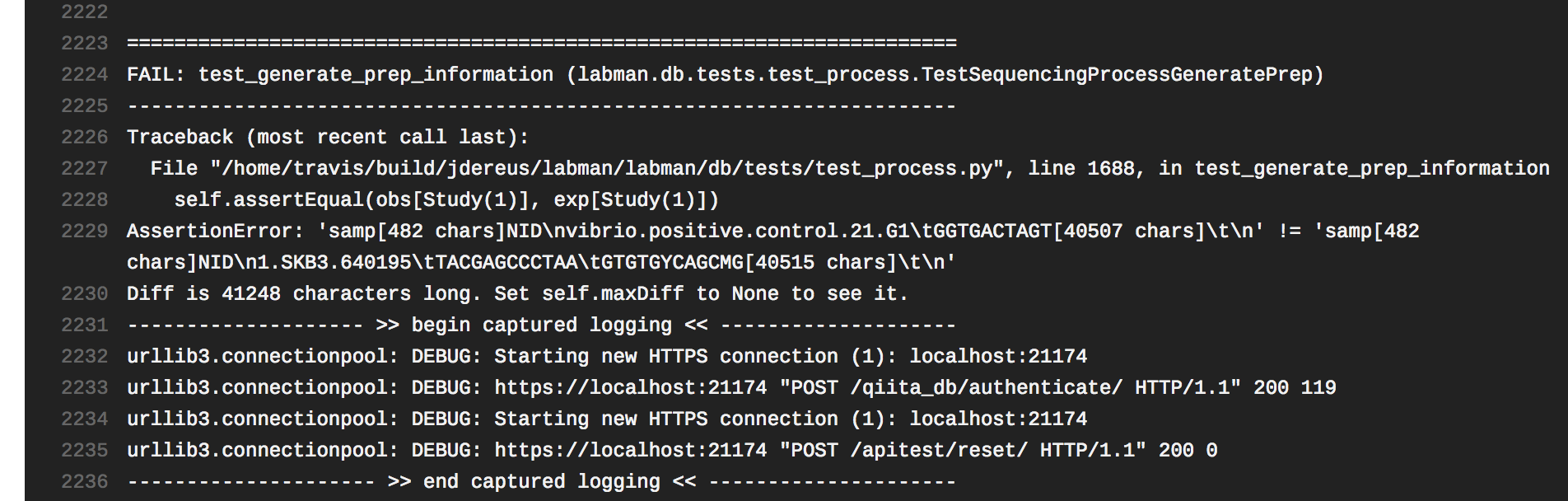
labman/db/process.py
Outdated
| data[study][content] = result | ||
| # if we want the sample_name.well_id, just replace sid | ||
| # for content | ||
| data[study][sid] = result |
There was a problem hiding this comment.
@antgonza I may have found your bug. Because the same sample can appear multiple times (even on a single plate, let alone on multiple plates throughout a sequencing run), if data is a dictionary of dictionaries (looks like it?) then the added line here,
data[study][sid] = result
... will overwrite info about those multiple samples each time the same sample id is encountered, leaving you with just one record for each sample (which, given that the SQL results aren't ordered, could be any one of them randomly--just whichever happens to be returned last).
The original code,
data[study][content] = result
would not be anticipated to have this problem because sid(sample_id) and content are not equivalent: content always (per code referenced below) has plate.id and well.well_id appended to it. Thus, in the case where a sample is plated multiple times, a single sample_id, say "1.SKB3.640195", ends up associated with multiple distinct contents such as "1.SKB3.640195.21.C9", "1.SKB3.640195.21.C12", etc, etc.
There was a problem hiding this comment.
@AmandaBirmingham and I discussed this offline and agreed on data[study][content] = result even so that this will generated duplicated sample names, which are not supported downstream.
There was a problem hiding this comment.
I also checked this one with Gail. She confirms that:
- The sample_name column must exactly match what is in Qiita (so we can't use sample content there, even if there are duplicates) and
- Every sample that is in the run must be represented in the prep info file (so, even if there are duplicates, we can't leave those out of the file)
* Note that while duplicate samples on the same plate or even in the same run is a very undesirable case in the real world, it is the norm in the test data. If you want to change the test db population and attendant unit tests so that they represent a more realistic case (no duplicate samples), that would be great!
In the case of duplicate samples in the same run, Gail manually goes into Qiita and adds a new sample (to represent the duplicate) and then manually modifies the name of the duplicate in the prep file to match the name she created for the new sample in Qiita. She has asked for Labman to show a warning when users download a prep info file that has duplicate samples in it, telling them that they will need to go through this process. I have added this as #343 Warn user if prep info file contains duplicate samples.
There was a problem hiding this comment.
Kewl, just note that in the current state we will have a difference in the blanks and spikes in the prep and the sample information file, which we will need to add to the sample info in Qiita somehow.
There was a problem hiding this comment.
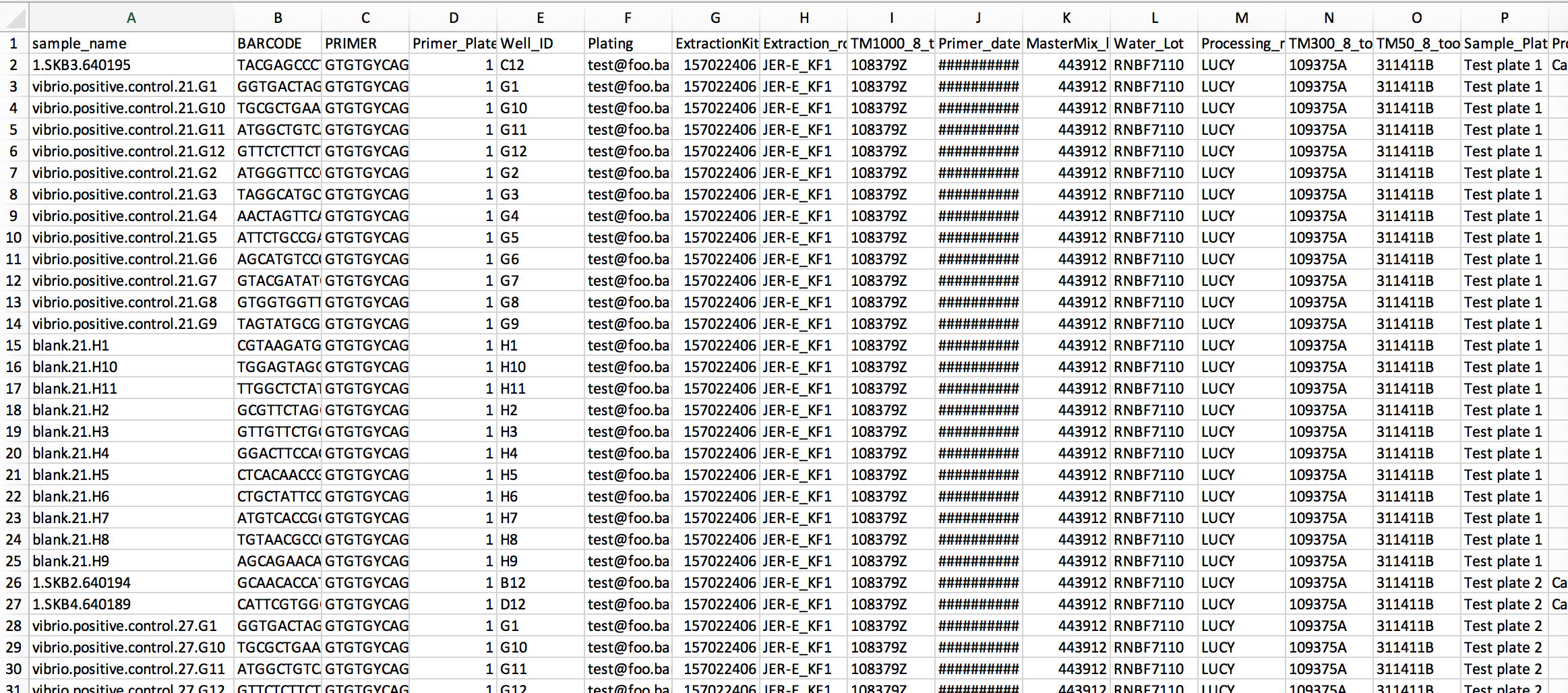
@antgonza You are absolutely right that the foregoing applies only to experimental samples--thanks for pointing that out! The known-good example we have for the prep info file shows that blanks are qualified by their well id while samples are not:
Furthermore, the labman.sample_composition table doesn't even store a sample_id for blanks and other controls:

Therefore, it looks like the sample_name column in the prep info file will need to contain the sample_id for experimental samples (i.e., those with sample_composition_type_id=1) and the content for everything else. Good catch!
There was a problem hiding this comment.
Related to @antgonza 's point above about blanks, etc: it looks like handling them requires a modification to our way of generating the Well_Description, too. The known-good file we got from the lab shows well descriptions like this:
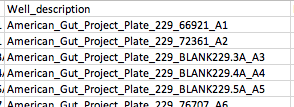
However, since as noted above the sample_composition table doesn't even store a sample_id for blanks and controls, following our approach of building the well description from the sample_id in these cases gives well descriptions with None in them, like this:
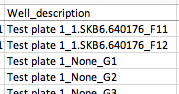
I'm coming to the conclusion that maybe everywhere in the prep info file generation that we incorporate sample_id for experimental samples (e.g., in sample name, well description, original name, more?), we need to instead incorporate content for the non-experimental samples to match the known-good. @antgonza , what do you think?
There was a problem hiding this comment.
I think this should be a case by case scenario:
- orig_name = name from qiita or empty for blanks/spikes
- sample_name = original name for unique samples, if samples are duplicated, use content; for blanks/spikes use content
- rest, like well description: use the previous sample name
This is an easy change in the current code; however, not easy to test as we will need to create a test with only unique sample ids and I have no idea of how to do that ...
There was a problem hiding this comment.
From the known-good we got from the lab, it looks like they currently expect orig_name to match sample_name even for blanks, etc (see below) so let's follow their convention on that. I agree with the suggestions for all the rest!
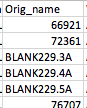
Re testing: Yeah, testing is going to be sticky :( The labman test db only knows about ONE Qiita study ("Identification of the Microbiomes for Cannabis Soils") and says that study only has 27 samples (not enough to make a full plate without either repetition or lots of blanks). It looks like this info comes from qiita/qiita_db/support_files/populate_test_db.sql , rather than from anything within labman, so I'm hesitant to try to modify its content in the labman db population scripts. I don't want to change the existing test plates in the db to not have any duplicate samples because we need to be able to test the behavior of full plates. Alternatively, adding new test plates that don't have duplicate samples on them would be a big effort in expanding the db population script; that effort should really be put in at some point, but it is going to be enough of a pain that I don't want to do it until such time as we have access to a larger test study in the qiita tables to "plate".
Given all this, I think the best of a host of bad options may be to change the db population script so that there are a small number of unique samples on the existing plates. This will be a pain and kind of hacky (and will require some changes to the existing unit tests--ugh!) but will mean that we will be able to test all the use cases (unique experimental samples, duplicated experimental samples, and blanks/etc) at least once. I can probably get a PR ready for that today.
There was a problem hiding this comment.
Note that it's pretty easy to create new studies in qiita via the qiita-api or even adding new samples to an existing study without having to modify qiita itself but this will need to be done via the qiita_client (which is part of of the db reset for tests).
There was a problem hiding this comment.
A PR that puts one unique sample on each of the sample plates in the test db (replacing one of the samples that was previously a repeat) is now available: #346 Test database extension: some unique samples
labman/db/process.py
Outdated
| -- orig_name | ||
| sample_id AS orig_name, | ||
| -- experiment_design_description | ||
| '' AS experiment_design_description, |
There was a problem hiding this comment.
The design description is always empty?
There was a problem hiding this comment.
...same question for some of those below
There was a problem hiding this comment.
Actually, this one is a mistake on my part--thanks for noticing it! I classed this in the "leave empty" category (see below) because it is empty in the known-good example I have, but going back to my notes, I see that Gail suggested that we should pull it from the Qiita study description.
Gail instructed us to always leave the following fields empty:
RUN_DATE
RUN_PREFIX
center_project_name
INSTRUMENT_MODEL
RUNID
I assume that someday in the future, labman or qiita or something will be expanded to close the loop by filling in these values after the sequencing is done ...
There was a problem hiding this comment.
Okay, thanks! Some of this is out-of-scope for labman too (e.g., the actual sequencing date)
There was a problem hiding this comment.
K, using qiita.study.study_description
labman/db/process.py
Outdated
| -- TM1000_8_tool | ||
| epmotion_tool_id, | ||
| -- mAStermix_lot | ||
| mASter_mix_id, |
There was a problem hiding this comment.
nop, changing ...
| -- experiment_design_description | ||
| '' AS experiment_design_description, | ||
| -- run_center | ||
| 'UCSDMI' AS run_center, |
There was a problem hiding this comment.
Is the center the same if we run on the miseq that sits in our lab space vs. the instruments that are part of the sequencing core? Or those at rady?
There was a problem hiding this comment.
When I asked "Is this always UCSDMI ?", the answer I got from Gail was "For our lab YES". I interpreted that to mean that it was true for all prep info sheets created in the lab; should I be asking her a more nuanced question here, and if so, what?
There was a problem hiding this comment.
Okay. I think it would be useful to ask Gail if UCSDMI encompasses any sequencing instrument associated with the UCSD campus. If yes, than we're good. If no, than more granularity may be warranted
There was a problem hiding this comment.
Ok, I've put a question out to Gail. If I learn that we need to make the behavior for this field more nuanced, I'll share that here.
There was a problem hiding this comment.
will go with UCSDMI, we can add an issue or replace once we have an answer if before merging ...
There was a problem hiding this comment.
Got an answer from Gail, which fortunately doesn't require a code change :)
Q: Is the center the same if we run on the miseq that sits in our lab space vs. the instruments that are part of the sequencing core? Or those at rady?
A: UCSDMI refers to all of those machines - in my opinion. Each machine has an identifier that is in the ruined allowing us to determine which particular site at UCSDMI was used (UCSD microbiome initiative).
fd183a3 to
fb45317
Compare
|
@AmandaBirmingham, just wondering if there is anything missing from this PR? Thanks! |
|
@antgonza It looks to me like we are still getting Nones incorporated in the Well_description for non-experimental samples like blanks and controls (see example in #328 (comment) ); as far as I know, that's the last remaining item! |
|
Good catch, fixing. |
No description provided.