| Author: | Andrew Tarzia |
|---|
A Monte Carlo-based molecular optimizer for optimizing the length of specified bonds in a molecule toward a target using cheap and unphysical potentials.
Please submit an issue with any questions or bugs!
Install using pip:
pip install mchammerSubunits are the rigid bodies of a molecule. Substructures are the bonded interactions between them. There are nonbonded interactions between subunits too.
MCHammer implements a simple Metropolis Monte-Carlo algorithm to optimize the
desired bonds toward a target bond length.
We define a graph of the molecule at the atomic level, which is further
coarse-grained into subunits that are separated by the bonds to be
optimized.
All atom positions/bond lengths within a subunit are kept rigid and do not
contribute to the potential energy, other than through nonbonded interactions.
The algorithm uses a simple Lennard-Jones nonbonded potential and parabolic
bond potential to define the potential energy surface such that the target
bond length is the energy mininum and steric clashes are avoided.
The MC algorithm is as follows:
- For
stepin N steps: - Choose a bond
Bat random. - Choose a subunit
son either side ofBat random.
3. Define two possible translations of
s,aandband choose at random:ais defined by a random number in [-1, 1) step along thesto molecule centre of mass (com).bis defined by a random number in [-1, 1) step along the vectorB. Step size is defined by user input.- Compute system potential
U=U_b+U_nb: U_bis the bonded potential, defined by the sum of all parabolic bond stretches about the target bond length for allB:U_b = sum_i (epsilon_b * (r_i - R_t)^2 ), whereR_tis the target bond length,epsilon_bdefines the strength of the potential andr_iis theithbond length.U_nbis the nonbonded potential, defined by the repulsive part of the Lennard-Jones potential:U_nb = sum_i,j (epsilon_nb * (sigma / r_ij)^mu), whereepsilon_nbdefines the strength of the potential,sigmadefines the position where the potential becomes repulsive,mudefines the steepness of the potential andr_ijis the pairwise distance between atomsiandj.
- Compute system potential
- Accept or reject move:
Accept if
U_i<U_(i-1)orexp(-beta(U_i - U_(i-1))>R, whereRis a random number [0, 1) andbetais the inverse Boltzmann temperature. Reject otherwise.
- Choose a bond
The workflow for a porous organic cage built using stk
(https://stk.readthedocs.io/) is shown schematically below (this example is
shown in examples/stk_example.py):
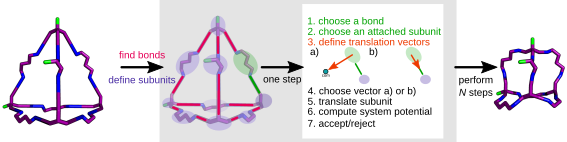
This code was originally written for use with stk (https://stk.readthedocs.io/), which assembles structures with long bonds that we wanted to optimize quickly. Now it has been generalized to take any molecule (defined by atoms and bonds) and a set of bonds to optimize to some target bond length. The algorithm is unphysical in that the bonded and nonbonded potential we apply is meaningless, other than to give a reasonable structure for further optimisation or use in a workflow!
The Optimizer class provides two main method: get_result and get_trajectory
, which provide the optimized structure and properties or the structure and
properties of each MC step, respectively.
The molecule at each step of the trajectory and its properties can be
collected from the results of the get_trajectory method, but not the
get_result method (the example file: examples/minimum_example.py shows how
to plot the optimisation progress and output the trajectory).
In this example, we use stk for I/O only with the input file available in
examples/minimum_example.py:
import stk
import mchammer as mch
benzene = stk.BuildingBlock.init_from_file('benzene.mol')
benzene_atoms = [
(atom.get_id(), atom.__class__.__name__)
for atom in benzene.get_atoms()
]
benzene_bonds = []
for i, bond in enumerate(benzene.get_bonds()):
b_ids = (bond.get_atom1().get_id(), bond.get_atom2().get_id())
benzene_bonds.append((i, b_ids))
mch_mol = mch.Molecule(
atoms=(
mch.Atom(id=i[0], element_string=i[1])
for i in benzene_atoms
),
bonds=(
mch.Bond(id=i[0], atom_ids=i[1])
for i in benzene_bonds
),
position_matrix=benzene.get_position_matrix(),
)
target_bond_length = 1.2
optimizer = mch.Optimizer(
step_size=0.25,
target_bond_length=target_bond_length,
num_steps=100,
)
subunits = mch_mol.get_subunits(
bond_pair_ids=((2, 3), (1, 5)),
)
# Get all steps.
mch_mol, mch_result = optimizer.get_trajectory(
mol=mch_mol,
bond_pair_ids=((2, 3), (1, 5)),
subunits=subunits,
)
# Update stk Molecule with new position matrix.
benzene = benzene.with_position_matrix(
mch_mol.get_position_matrix()
)
benzene.write('benzene_opt.mol')Finally, we mention that the Optimizer.get_subunits() is based on splitting
the Molecule by the input bond_pair_ids.
This method is now public, so that users can modify the defined subunits to
enforce rigid non-covalent interactions.
I.e. non-covalent complexes will be distinct subunits because there is no bond
between them, and the user can merge them into one subunit by merging the
iterable of atom ids in the subunits dictionary, to force the algorithm to
treat them as one rigid body.
An example of this is given in examples/stk_example.py using an arbitrary
non-covalent complex BuildingBlock.
As part of this code, I also provide the Collapser class, which is a naive
precursor to MCHammer that simply moves all subunits toward the molecule
centroid until a distance threshold is met.
This can sometimes be faster than MCHammer for some molecule types.
An example of this is shown in examples/collapser_example.py.
I developed this code as a post doc in the Jelfs research group at Imperial College London (<http://www.jelfs-group.org/>, <https://github.com/JelfsMaterialsGroup>).
This code was reviewed and edited by: Lukas Turcani (<https://github.com/lukasturcani>), Steven Bennett (<https://github.com/stevenbennett96>)
This project is licensed under the MIT license.