SeqFindr - easily create informative genomic feature plots. It's a bioinfomagicians tool to detect the presence or absence of genomic features given a database describing these features & a set of draft and/or complete genomes. We work with bacterial genomes & as such SeqFindr has only been tested with bacterial genomes.
I am on vacation from 08/12/14 -> 06/01/15. User support will not happen in this period. Stupidly I've done some releases the day/night before I leave.
If you have problems:
$ # (sudo if neded) $ pip uninstall SeqFindr $ # Run the above command numerous times to ensure it's gone # pip install SeqFindr==0.33.1


Please use this README.rst as the core SeqFindr user documentation.
- These are works in progress:
07/12/14: I am on vacation from 08/12/14 -> 06/01/15. User support will not happen in this period. Stupidly I've done some releases the day/night before I leave.
If you have problems:
$ # (sudo if neded) $ pip uninstall SeqFindr $ # Run the above command numerous times to ensure it's gone # pip install SeqFindr==0.33.1
18/11/14: Version 0.4.0 now has new option --remove_empty_cols. It will strip out entire columns where no hits were detected.
28/07/14: Fixed a bug where axes were shifted when using newer versions of matplotlib.
Important: Were you using a specific SeqFindr version as a dependency for you project and it has disappeared from PyPI?
We recently activated a name change of SeqFind*R* to SeqFind*r*. This was to avoid potential users believing this was a R package. Unfortunately, PyPI while aware that SeqFindR and SeqFindr were different packages did not like the potential confusion. As a consequence the only resolution was to delete SeqFind*R* completely (and losing all PyPI published releases) and registering SeqFind*r* and starting fresh. All previous 10 releases, while not available on PyPi are still available on GitHub. If you require a previous release you can actually do something like this (SeqFindr v0.26):
pip install -e git://github.com/mscook/SeqFindr.git@v0.26
Version 0.31.1 released on 10 July 2014.
We are now testing SeqFindr builds on both Linux & MacOSX systems.
Best use "git log" for a changelog as the changelog for most recent changes/fixes/enhancements may not be up to date.
Cite this Github repository if you use SeqFindr to generate figures for publications:
STANTON-COOK M, NF ALIKHAN, FORDE BM, BEN ZAKOUR NL & BEATSON SA^. SeqFindr - easily create informative genomic feature plots. https://github.com/mscook/SeqFindr.
TODO: Couple SeqFindr with ZENODO!
SeqFindr is a commandline application. If you're not familiar with the commandline we recommend you ask local IT support to help you install it.
We now test SeqFindr builds on both Linux (Ubuntu >= 12.04) and MacOSX (Mavericks) systems.
- You will need to install/have installed:
- ncbiblast >= 2.2.27
- python >= 2.7 (Python 3 is not supported)
You can check these are installed by:
$ python --version $ blastn -version
Installation of python or blastn (without a package manager) is beyond the scope of this document.
If you have both python and blastn you need to (if not already present) install pip.
You can check if pip exists with:
$ which pip
If you get a "not found", please read the pip installation instructions.
If you already have pip we do suggest you upgrade it. We are using version 1.5.6 at the time of writing this document.
You can upgrade pip like this:
$ pip install --upgrade pip
The following python libraries should be installed (automatically) if you follow the installation instructions detailed below.
- We use the following python libraries:
- numpy >= 1.6.1
- scipy >= 0.10.1
- matplotlib >= 1.1.0
- biopython >= 1.59
- ghalton>=0.6
These libraries will also have dependencies (i.e. atlas, lapack, fortran compilers, freetype and png). These most likely won't be installed on your computer. Please install these before attempting the installation.
SeqFindr uses 3rd party packages that are extremely important for scientific computing but are notoriously difficult to install. While pip install * *--user SeqFindr may work we recommend you install these 3rd party packages using apt-get.
Run:
$ sudo apt-get install python-numpy python-scipy python-matplotlib python-biopython python-dev libatlas-dev liblapack-dev gfortran libfreetype6-dev libfreetype6 libpng-dev
Now pip install SeqFindr:
$ pip install --user SeqFindr
We use the --user option of pip to put SeqFindr in: /home/$USER/.local/bin/ You need to add this location to you ~/.bash_profile.
Add SeqFindr to your path:
$ echo 'export PATH=$PATH:/home/$USER/.local/bin/' >> ~/.bash_profile
Finally install BLAST+:
$ sudo apt-get install ncbi-blast+
Test it:
Run:
$ SeqFindr -h $ python -c 'import SeqFindr; print SeqFindr'
You'll need to have the equivalents of python-dev libatlas-dev liblapack-dev gfortran libfreetype6-dev libfreetype6 & libpng-dev installed. We had no problems installing SeqFindr on a recently acquired OSX Mavericks machine using the homebrew package manager.
The installed packages on this machine via:
$ brew list
Are available at this gist.
pip install SeqFindr:
$ pip install --user SeqFindr
We use the --user option of pip to put SeqFindr in: /home/$USER/.local/bin/ You need to add this location to you ~/.bash_profile.
Add SeqFindr to your path:
$ echo 'export PATH=$PATH:/home/$USER/.local/bin/' >> ~/.bash_profile
Finally install BLAST+:
$ sudo brew install blast
Test it:
Run:
$ SeqFindr -h $ python -c 'import SeqFindr; print SeqFindr'
You can upgrade like this:
pip install --upgrade SeqFindr
Please regularly check back to make sure you're running the most recent SeqFindr version.
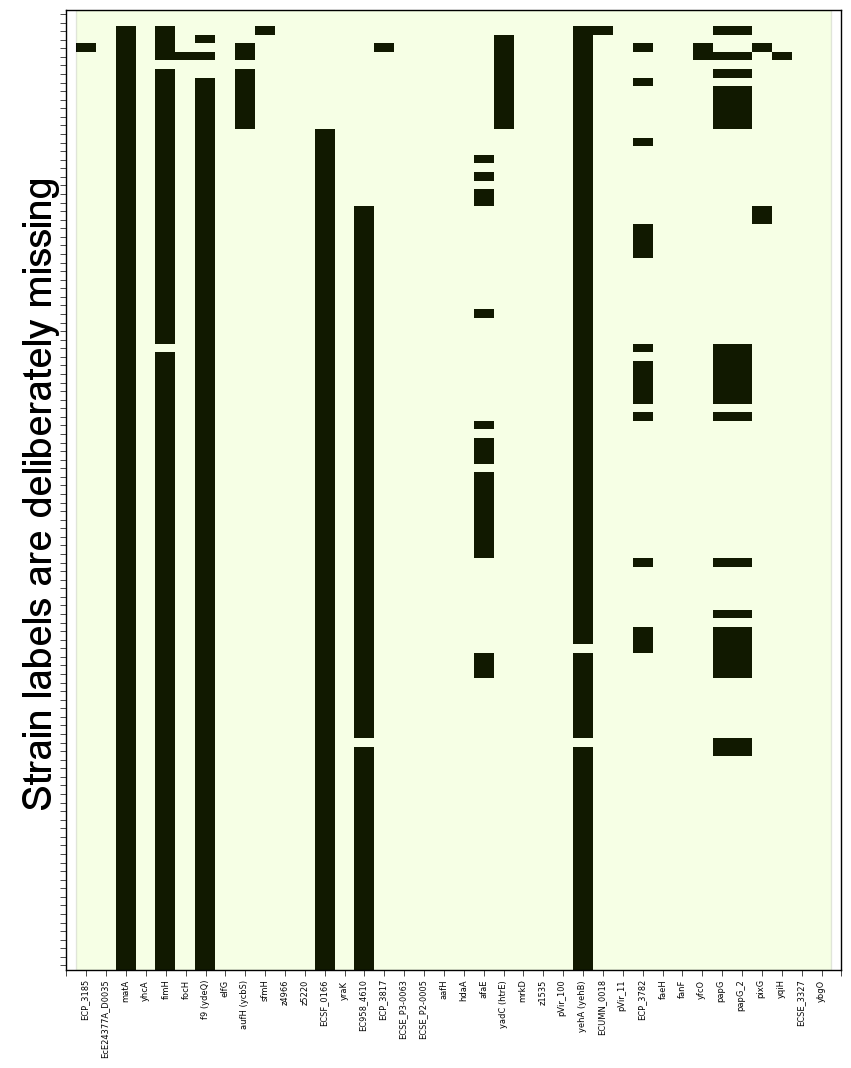
SeqFindr CU fimbriae genes image. 110 E. coli strains were investigated. Order is according to phylogenetic analysis. Black blocks represent gene presence.
The SeqFindr database is in multi-fasta format. The header needs to be formatted with 4 comma separated elements. We concede that inventing another file format is annoying, but, future versions of SeqFindr will exploit this information.
- The elements headers are:
- identifier,
- common name (this is taken as the gene label in the plot),
- description and
- species
The final element, separated by [] contains a classification. This information is used by SeqFindr to draw different coloured blocks.
An example:
>70-tem8674, bla-TEM, Beta-lactams Antibiotic resistance (ampicillin), Unknown sp. [Beta-lactams] AAAGTTCTGCTATGTGGCGCGGTATTATCCCGTGTTGACGCCGGGCAAGAGCAACTCGGTCGCCGCATAC >70-shv86, bla-SHV, Beta-lactams Antibiotic resistance (ampicillin), Unknown sp. [Beta-lactams] CTCAAGCGGCTGCGGGCTGGCGTGTACCGCCAGCGGCAGGGTGGCTAACAGGGAGATAATACACAGGCGA >70-oxa(1)256, bla-OXA-1, Beta-lactams Antibiotic resistance (ampicillin), Unknown sp. [Beta-lactams] >70-tetB190, tet(B), Tetracycline Antibiotic resistance (tetracycline), Unknown sp. [Tetracycline] CAAAGTGGTTAGCGATATCTTCCGAAGCAATAAATTCACGTAATAACGTTGGCAAGACTGGCATGATAAG
Note: if you do not have all information you can simplify the expected database header to:
>, bla-TEM, , [classification]
The script vfdb_to_seqfindr is now included in SeqFindr to convert VFDB formatted files (or like) to SeqFindr formatted database files.
VFDB: Virulence Factors Database (www.mgc.ac.cn/VFs/) is a reference database for bacterial virulence factors.
At this stage we have tested this script on limited internal datasets. Success/mileage will depend on the consistency of the VFDB formatting.
Example usage of vfdb_to_seqfindr:
# Default (will set VFDB classification identifiers as the classification) $ vfdb_to_seqfindr -i TOTAL_Strep_VFs.fas -o TOTAL_Strep_VFs.sqf # Sets any classification to blank ([ ]) $ vfdb_to_seqfindr -i TOTAL_Strep_VFs.fas -o TOTAL_Strep_VFs.sqf -b # Reads a user defined classification. 1 per in same order as input # sequences $ python convert_vfdb_to_SeqFindr.py -i TOTAL_Strep_VFs.fas -o TOTAL_Strep_VFs.sqf -c user.class
The -c (--class_file) option is very useful. Suppose you want to annotate your sequences of interest with user defined classification values. Simply develop a file containing the scheme as pass using the -c option (3rd example above). A sample file for the situation where you had 7 input sequences with the first 3 Fe transporters, the next two Toxins, the next a Misc and the final sequence is a Toxin would look like this:
Fe transporter Fe transporter Fe transporter Toxin Toxin Misc Toxin
We use the following calculation:
hsp.identities/float(record.query_length) >= tol
- Where:
- hsp.identities is number of identities in the high-scoring pairs between the query (database entry) and subject (contig/scaffold/mapping consensus),
- record.query_length is the length of the database entry and,
- tol is the cutoff threshold to accept a hit (0.95 default)
For a database entry of 200 bp you can have up to 10 mismatches/gaps without being penalised.
- Why not just use max identity?
- Eliminate effects of scaffolding characters/gaps,
- Handle poor coverage etc. in mapping consensuses where N characters/gaps may be introduced
What problems may this approach cause? I'm still looking into it...
SeqFindr can read a configuration file. At the moment you can only redefine the category colors (suppose you want to use a set of fixed colors instead of the default randomly generated). The configuration file is expected to expand in the future.
To define category colors:
touch ~/.SeqFindr.cfg vi ~/.SeqFindr.cfg # Add something like category_colors = [(100,60,201), (255,0,99)]
Category colors can be any RGB triplet. You could use a tool similar to this one: http://www.colorschemer.com/online.html
For example the first row of colors in RGB is: (51,102,255), (102,51,255), (204,51,255), (255,51,204)
In some cases you may want to screen using PCR primers. Please use the --short option. Here we adjust BLASTn parameters wordsize = 7 & Expect Value = 1000
We provide a script to run all the examples. Note: We have changed the color generation code. As a consequence the background colors will be different when running this yourself. The results will not change.
- Navigate to the SeqFindr/example directory (from git clone). The following files should be present:
- A database file called Antibiotic_markers.fa
- An ordering file called dummy.order (-i option)
- An assemblies directory containing strain1.fa, strain2.fa and strain3.fa
- A consensus directory containing strain1.fa, strain2.fa and strain3.fa (-m option)
- Note: the assembly and consensus directories contain:
- the same number of files (3 each)
- there is a 1-1 filename mapping (strain1.fa, strain2.fa, strain3.fa == strain1.fa, strain2.fa, strain3.fa)
- there are only fasta files. If you wish to include complete genomes either download the genomes in fasta format OR convert the Genbank or EMBL files to fasta format.
- The toy assemblies and consensuses were generated such that:
- strain1 was missing: 70-shv86, 70-ctx143 and 70-aac3(IV)380 with mis-assembly of 70-aphA(1)1310 & 70-tem8674
- strain2 was missing: 70-oxa(7)295, 70-pse(4)348 70-ctx143, 70-aadA1588, 70-aadB1778 and 70-aacC(2)200
- strain2 was missing 70-shv86, 70-ctx143 and 70-aac3(IV)380 with mis-assembly of 70-aphA(1)1310, 70-tem8674 and 70-aadA1588
Something like this:
$ # Assuming you git cloned, python setup.py install $ cd SeqFindr/example $ ./run_examples.sh $ # See directories run1/ run2/ run3/ run4/
Command:
SeqFindr Antibiotic_markers.fa assemblies/ -o run1 -l
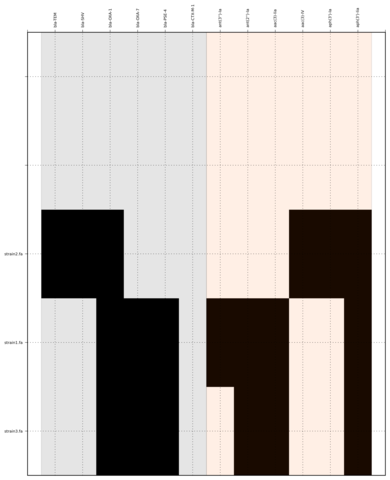
Link to full size run1.
Command:
SeqFindr Antibiotic_markers.fa assemblies/ -m consensus/ -o run2 -l
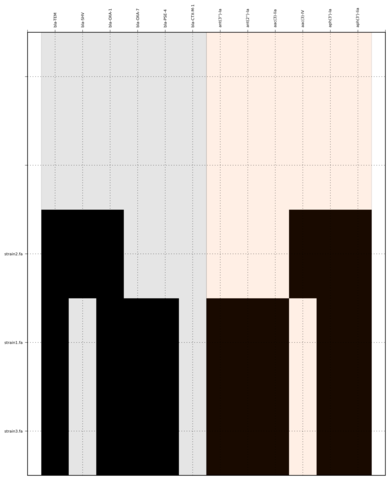
Link to full size run2.
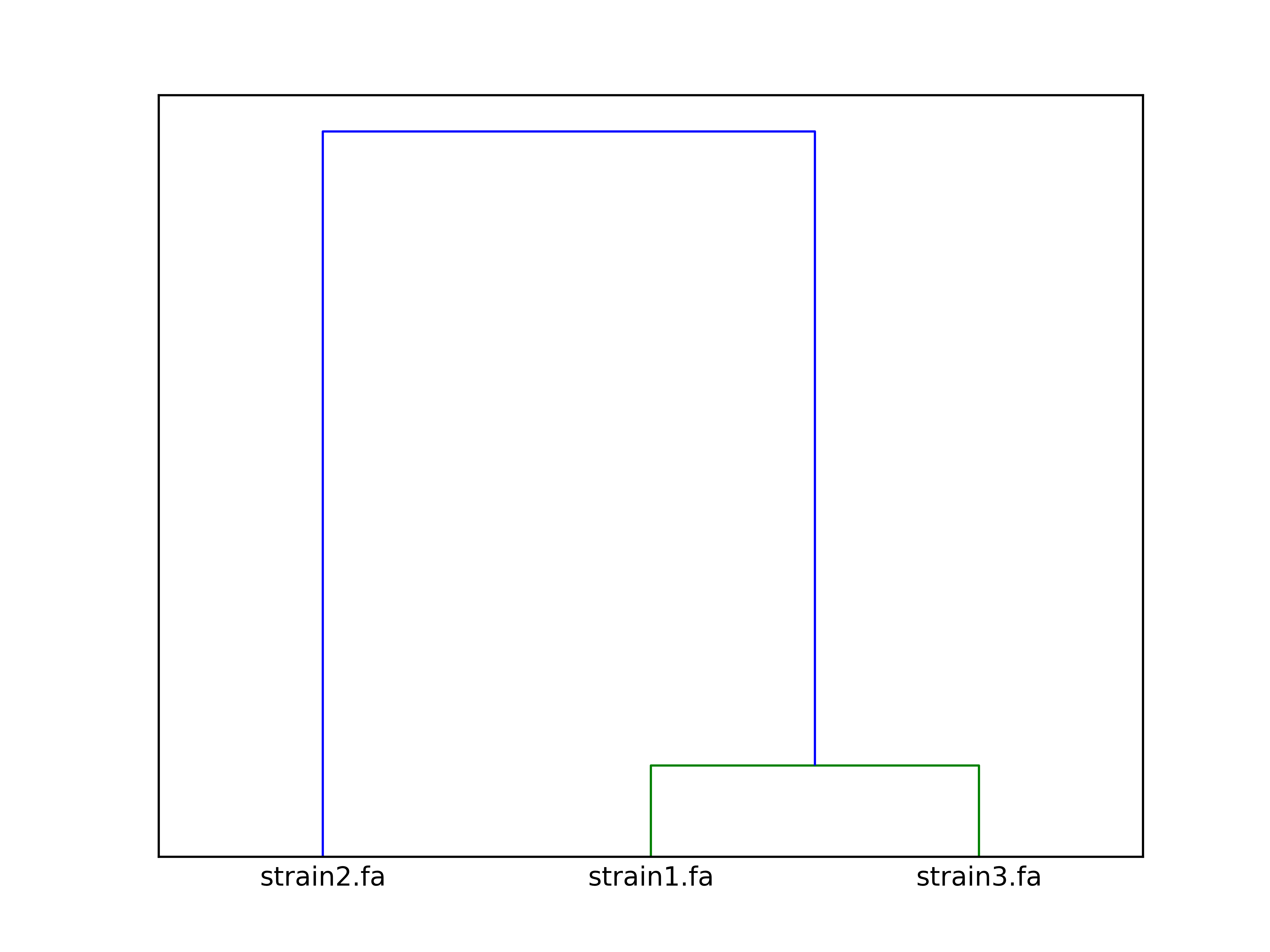
Command:
SeqFindr Antibiotic_markers.fa assemblies/ -m consensus/ -o run3 -l -r
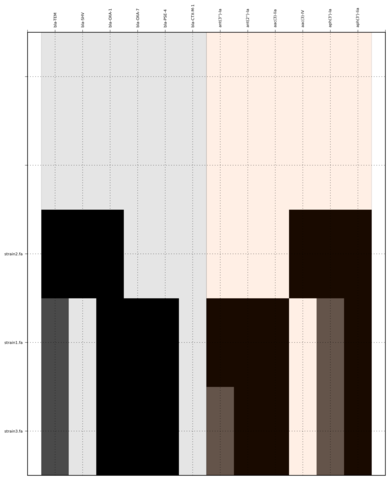
Link to full size run3.
The clustering dendrogram looks like this:
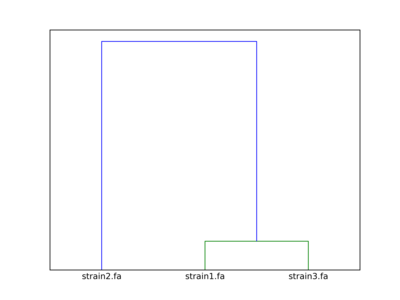
Link to full size dendrogram.
Note: the ordering file is defined using the option --index_file. The ordering file must contain the same number of strains as the assemblies directory and the strain names must agree (TODO - add a script to flag issues).
Command:
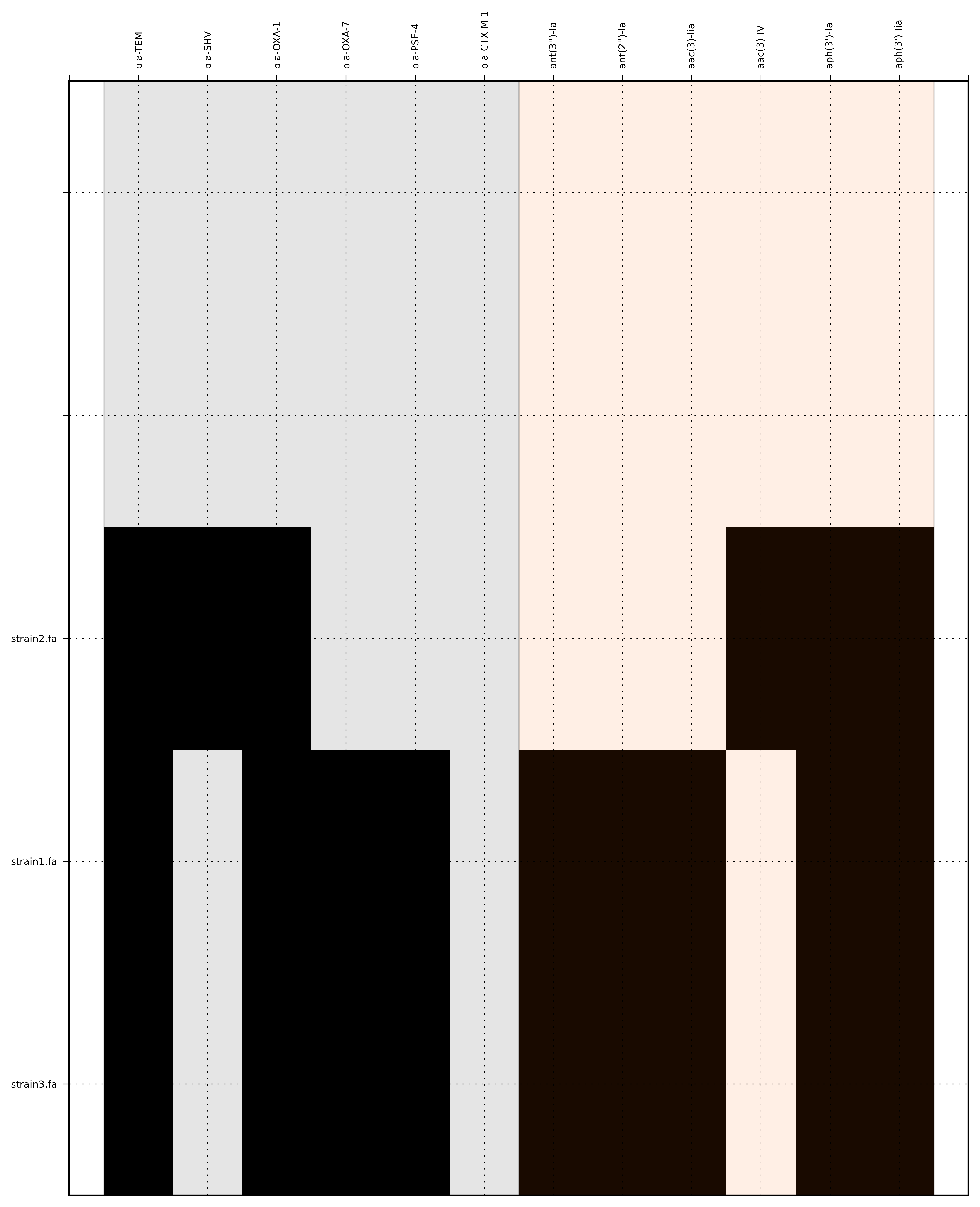
SeqFindr Antibiotic_markers.fa assemblies/ -m consensus/ -o run4 -l -r --index_file dummy.order
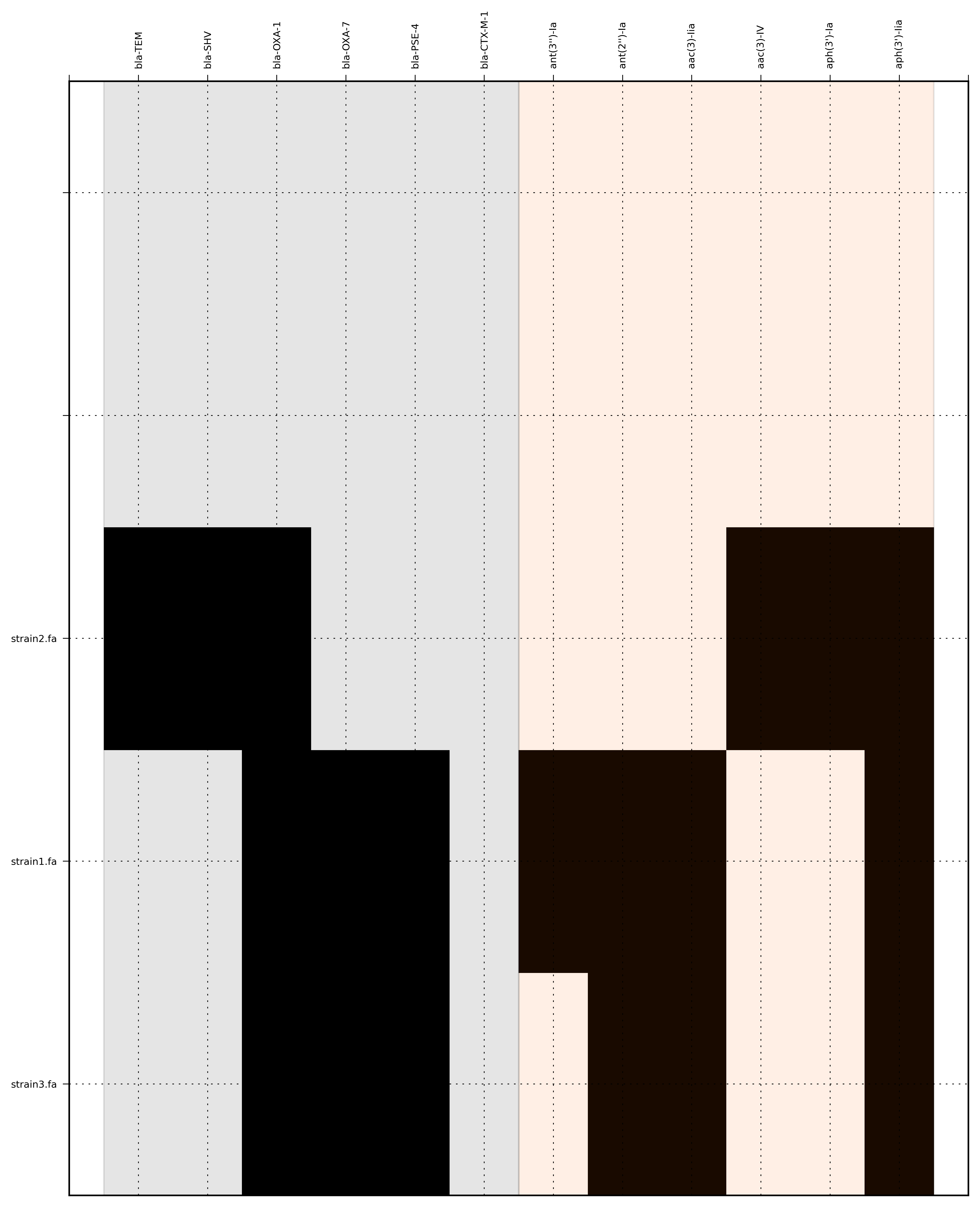{kind=link}
{kind=link}
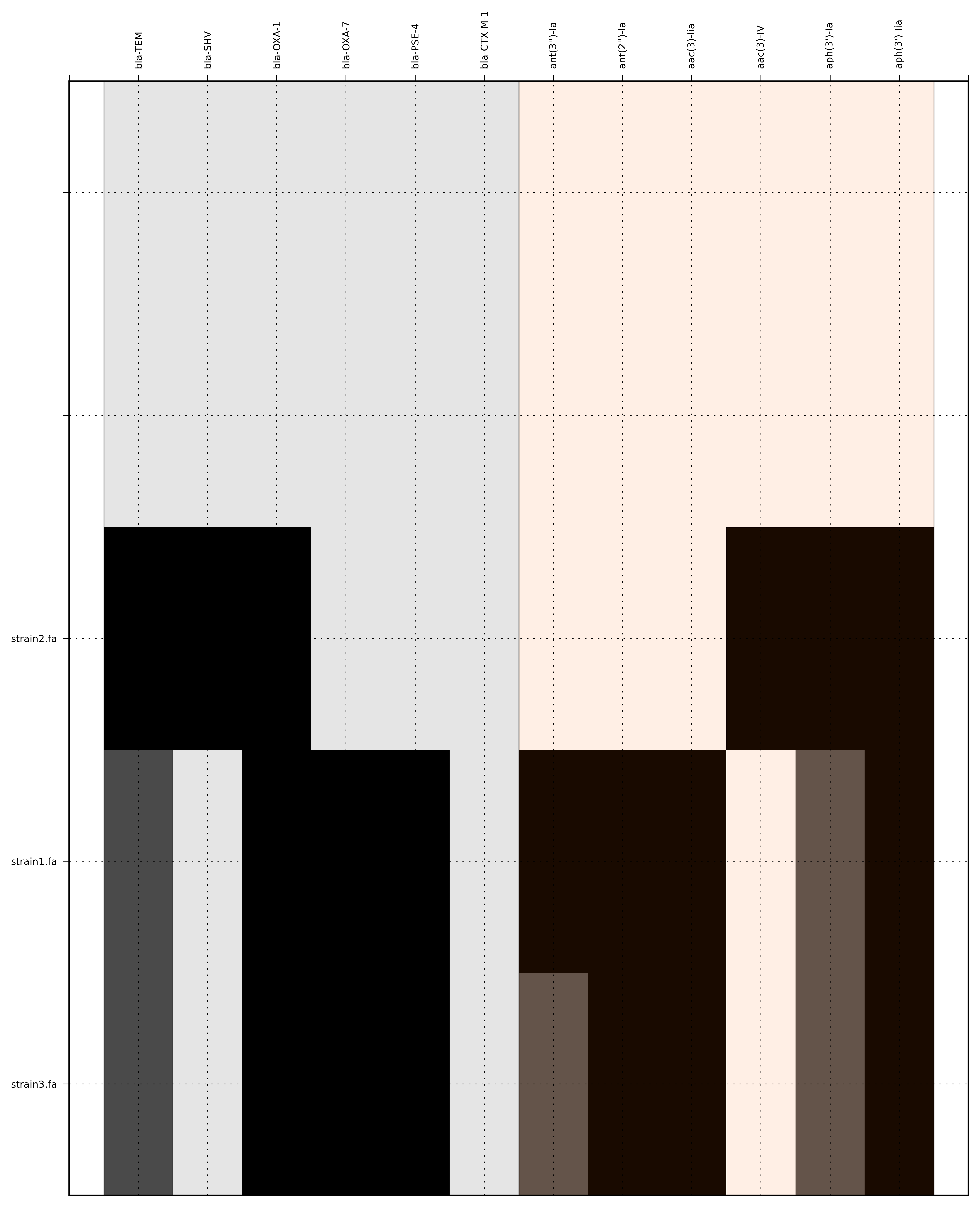{kind=link}
{kind=link}
Link to full size run4.
{kind=link}
We strongly recommend that you use mapping consensus data. It minimises the effects of missassembly and collapsed repeats.
We use Nesoni. We use the database file (in multi-fasta format) as the reference for mapping. Nesoni has no issues with multifasta files as references (BWA will treat them as separate chromosomes). The workflow is something like this:
$ nesoni make-reference myref ref-sequences.fa $ # for each strain $ # nesoni analyse-sample: mysample myref pairs: reads1.fastq reads2.fastq $ # extract the consensus.fa file
For those of you using a cluster running PBSPro see: https://github.com/mscook/SeqFindr_nesoni This is a script that generates a job array, submits and cleans up the mapping results ready for input to SeqFindr.
The output from the described workflow and SeqFindr_nesoni is a consensus.fa file which we term the mapping consensus. This file is a multi-fasta file of the consensus base calls relative to the database sequences.
- Caveats:
- you will probably want to allow multi-mapping reads (giving --monogamous no --random yes to nesoni consensus) (this is default for SeqFindr_nesoni),
- The (poor) alignment of reads at the start and the end of the database genes can result in N base calls. This can result in downstream false negatives.
SeqFindr now provides a solution to minimise the effects of poor mapping at the start and end of the given sequences.
The SeqFindr option is -s or --STRIP:
-s STRIP, --strip STRIP Strip the 1st and last N bases of mapping consensuses & database [default = 10]
By default this strips the 1st and last 10 bases from the mapping consensuses. We have had good results with this value. Feel free to experiment with different values (say, -s 0, -s 5, -s 10, -s 15). Please see image-compare a script we developed to compare the effects of different values of -s on the resultant figures.
See the help listing. You can get this yourself with:
$ SeqFindr -h
Please see the TODO for future SeqFindr project directions.