- FastQC 0.11.9+
- FeatureCounts 2.0.0+
- MultiQC 1.7+
- Python 3.6+ (using Ana(mini)conda)
- Samtools 1.3+
- Snakemake 5.7+
- SRA Toolkit 2.9.6+
- Trimmomatic 0.39+
- calibrate [http://cran.r-project.org/package=calibrate]
- DESeq2 [https://bioconductor.org/packages/3.10/bioc/html/DESeq2.html]
- dplyr [http://cran.r-project.org/package=dplyr]
- GGally [http://cran.r-project.org/package=GGally]
- ggplot2 [http://cran.r-project.org/package=ggplot2]
- ggrepel [http://cran.r-project.org/package=ggrepel]
- gplots [http://cran.r-project.org/package=gplots]
- gridExtra [http://cran.r-project.org/package=gridExtra]
- kableExtra [http://cran.r-project.org/package=kableExtra]
- knitr [http://cran.r-project.org/package=knitr]
- latex2exp [http://cran.r-project.org/package=latex2exp]
- pander [http://cran.r-project.org/package=pander]
- RColorBrewer [http://cran.r-project.org/package=RColorBrewer]
- rmarkdown [http://cran.r-project.org/package=rmarkdown]
git clone https://github.com/villegar/doge
cd doge
conda env create -f environment.yml -n DoGE
conda activate DoGE or source activate DoGE
python download.genome.py genomes/X-genome.jsonA good place to get some reference genomes and gene annotations is http://uswest.ensembl.org/info/data/ftp/index.html. The reference must be stored in JSON format (see below template),X-genome.json
{
"X.fa.gz":
"ftp://ftp.ensembl.org/pub/some/path/to/X.fa.gz",
"X.gtf.gz":
"ftp://ftp.ensembl.org/pub/some/path/to/X.gtf.gz"
}snakemake -j CPUS \ # maximum number of CPUs available to Snakemake
--configfile config.json # configuration filesnakemake -j JOBS \ # maximum number of simultaneous jobs to spawn
--configfile config.json # configuration file
--latency-wait 1000 \ # files latency in seconds
--cluster-config cluster.json \ # cluster configuration file
--cluster "sbatch --job-name={cluster.name}
--nodes={cluster.nodes}
--ntasks-per-node={cluster.ntasks}
--output={cluster.log}
--partition={cluster.partition}
--time={cluster.time}"bash run_cluster config.json &> log &{
"__default__" :
{
"time" : "1-00:00:00",
"nodes" : 1,
"partition" : "compute",
"ntasks": "{threads}",
"name": "DoGE-{rule}",
"log": "DoGE-{rule}-%J.log"
}
}- The
genomesection MUST point to the path for theX-genome.jsonfile. - The
readssection points the pipeline to the location (path), format (extension), type (end_type), and prefix (prefix) of the raw reads. Optionally, ifend_type = pe(paired-end), both the forward (forward_read_id) and reverse (reverse_read_id) reads identifier (e.g. 1, R1, 2, R2, etc.) should be specified. - The
trimmomaticsection should contain a sub-key calledoptionswith the parameters for trimming, excluding the input and ouput names, which will be set up by the pipeline.
{
"genome": "/path/to/X-genome.json",
"reads": {
"extension": "fastq",
"end_type": "se",
"forward_read_id": "1",
"reverse_read_id": "2",
"path": "/path/to/raw/reads",
"prefix": "SRR"
},
"trimmomatic":{
"options": "ILLUMINACLIP:{input.adapter}/TruSeq3-SE-2.fa:2:30:10:2:keepBothReads"
}
}For this study case the following article title LncRNA DEANR1 facilitates human endoderm differentiation by activating FOXA2 expression was consulted.
https://doi.org/10.1016/j.celrep.2015.03.008
SRR1958165
SRR1958166
SRR1958167
SRR1958168
SRR1958169
SRR1958170
{
"genome": "genomes/human-genome.json",
"reads": {
"extension": "fastq",
"end_type": "se",
"path": "/path/to/reads",
"prefix": "SRR"
},
"trimmomatic":{
"options": "ILLUMINACLIP:{input.adapter}/TruSeq3-SE-2.fa:2:30:10:2:keepBothReads TRAILING:3 MINLEN:24"
}
}
It is a good practice to perform a dry-run of the workflow before submitting for execution. This can be done by appending the -n option to the snakemake command:
snakemake --configfile config.json -n
The output will display a summary of each job that will be processed and a final summary that should look like:
Job counts:
count jobs
6 alignment
6 alignment_quality
1 all
1 annotation_table
6 fastqc_raw
6 fastqc_trimmed
1 feature_counts
1 hisat2_index
1 quantification_table
1 rmd_report
6 sam2bam
6 trim_reads
42
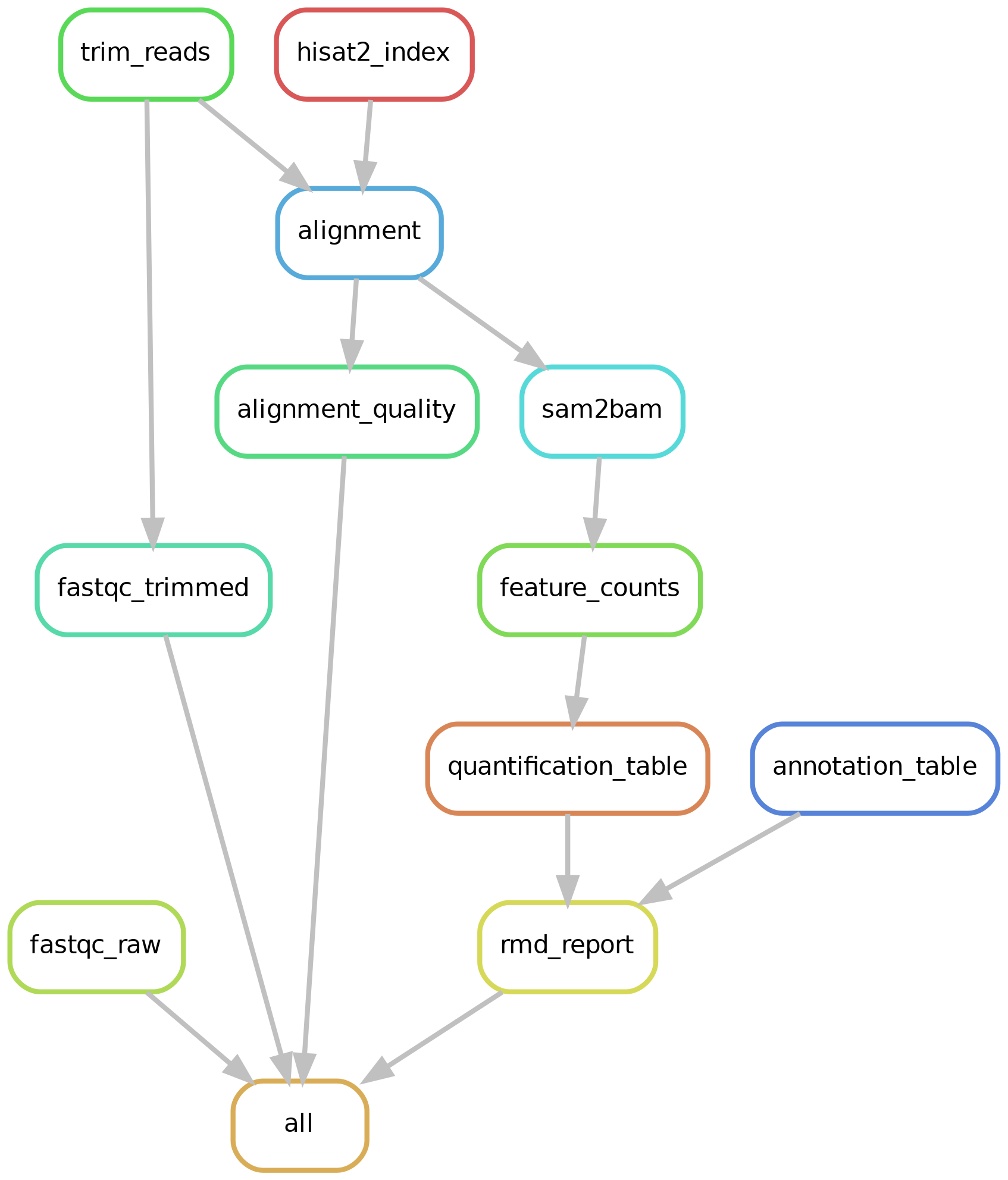
For a graphical summary of above jobs, check the directed acyciclic graph: https://raw.githubusercontent.com/villegar/DoGE/master/images/dag.png
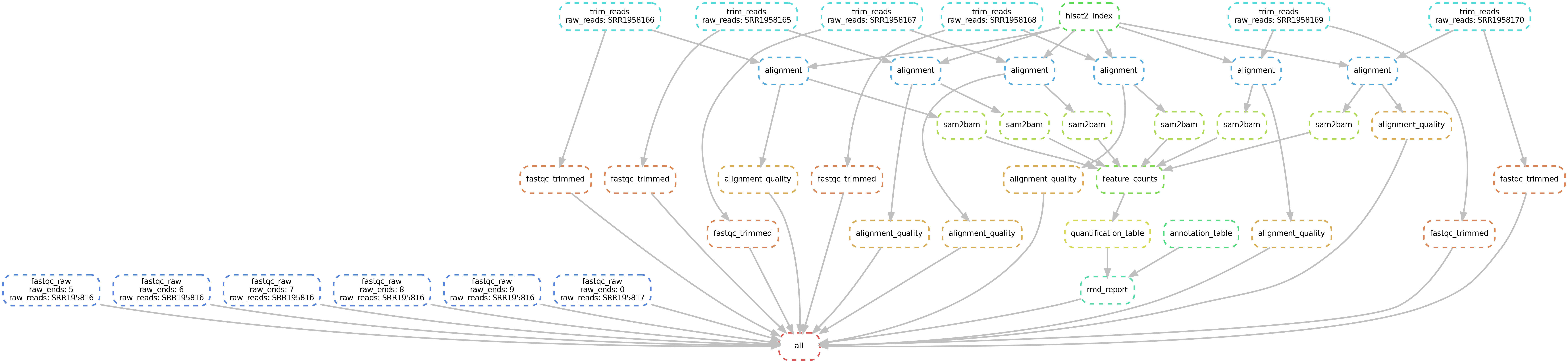{kind=link}
snakemake -j CPUS \ # maximum number of CPUs available to Snakemake
--configfile config.json # configuration file