# Loading Packages
library(ggPlantmap)
library(tidyverse)## ── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ──
## ✔ dplyr 1.1.4 ✔ readr 2.1.4
## ✔ forcats 1.0.0 ✔ stringr 1.5.0
## ✔ ggplot2 3.4.4 ✔ tibble 3.2.1
## ✔ lubridate 1.9.3 ✔ tidyr 1.3.0
## ✔ purrr 1.0.2
## ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
## ✖ dplyr::filter() masks stats::filter()
## ✖ dplyr::lag() masks stats::lag()
## ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errors
As you will notice, data organization is not standardized. Each study structures their quantitative data in their own specific way. Therefore, it is important that each specific dataset to be handle uniquely so that can be used to feed a ggPlantmap pipeline. Here, I present an example of a single-cell RNA-seq dataset from this paper:
Denyer, T., Ma, X., Klesen, S., Scacchi, E., Nieselt, K., & Timmermans, M. (2019). Spatiotemporal Developmental Trajectories in the Arabidopsis Root Revealed Using High-Throughput Single-Cell RNA Sequencing. Developmental cell, 48(6), 840–852.e5.
The datasets used for this guide can be found in the data folder
The data used for this guide was extracted from The Arabidopsis Root scRNA-Seq Atlas. We downloaded the data that shows the expression of four genes (AT1G03550, AT1G09750, AT1G29950 and AT5G06200) in the Arabidopsis Root with protoplasting-induced genes dataset.
data <- read.csv("data/download_avg_UMI.csv")
print(data)## X C0 C0. C1 C1. C2 C2. C3 C3. C4 C4. C5 C5.
## 1 AT1G03550 0.10 11.41 0.12 11.46 0.07 7.11 0.03 4.87 0.66 74.36 0.05 8.66
## 2 AT1G09750 0.00 0.38 0.00 0.19 0.00 0.22 0.04 6.64 0.00 1.17 0.00 0.50
## 3 AT1G29950 0.05 5.13 0.04 4.08 0.06 6.47 0.34 34.96 0.01 5.36 0.38 37.38
## 4 AT5G06200 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.88 0.00 1.63 0.01 0.99
## C6 C6. C7 C7. C8 C8. C9 C9. C10 C10. C11 C11. C12 C12.
## 1 0.04 10.03 0.02 7.46 0.04 11.51 0.02 3.23 0.02 5.20 0.01 2.74 0.10 10.49
## 2 0.00 1.94 0.01 1.36 0.01 1.19 0.00 0.00 0.00 0.58 1.39 75.34 0.01 1.40
## 3 0.02 8.74 0.03 10.85 0.01 3.97 0.03 5.91 0.04 8.67 0.00 0.68 0.26 23.78
## 4 0.00 0.32 0.00 1.69 0.00 1.59 0.00 1.61 0.32 2.89 0.00 0.00 0.00 0.70
## C13 C13. C14 C14. C15 C15. C16 C16.
## 1 0.02 2.19 0.01 4.0 1.19 80.65 0.06 7.69
## 2 0.00 0.00 0.00 0.8 0.00 0.00 0.00 0.00
## 3 1.48 71.53 0.03 6.4 0.00 0.00 0.03 5.13
## 4 0.00 0.00 0.00 1.6 0.00 0.00 0.00 0.00
The data is organized with gene names in the first column followed by the average expression (Raw UMI reads) for all the cells in a specific cluster (C0 to C16). Note that there is an additional column for each cluster (example: C0.), that describes the percentage of cells in the cluster that expresses the gene at a certain threshold. For now, we will only focus in the average expression values.
processed.data <- data %>%
pivot_longer(-X) %>% ## making it tidy
filter(!str_detect(name,"[.]")) ## removing columns that correspond to the percentage of cells with signal
processed.data## # A tibble: 68 × 3
## X name value
## <chr> <chr> <dbl>
## 1 AT1G03550 C0 0.1
## 2 AT1G03550 C1 0.12
## 3 AT1G03550 C2 0.07
## 4 AT1G03550 C3 0.03
## 5 AT1G03550 C4 0.66
## 6 AT1G03550 C5 0.05
## 7 AT1G03550 C6 0.04
## 8 AT1G03550 C7 0.02
## 9 AT1G03550 C8 0.04
## 10 AT1G03550 C9 0.02
## # ℹ 58 more rows
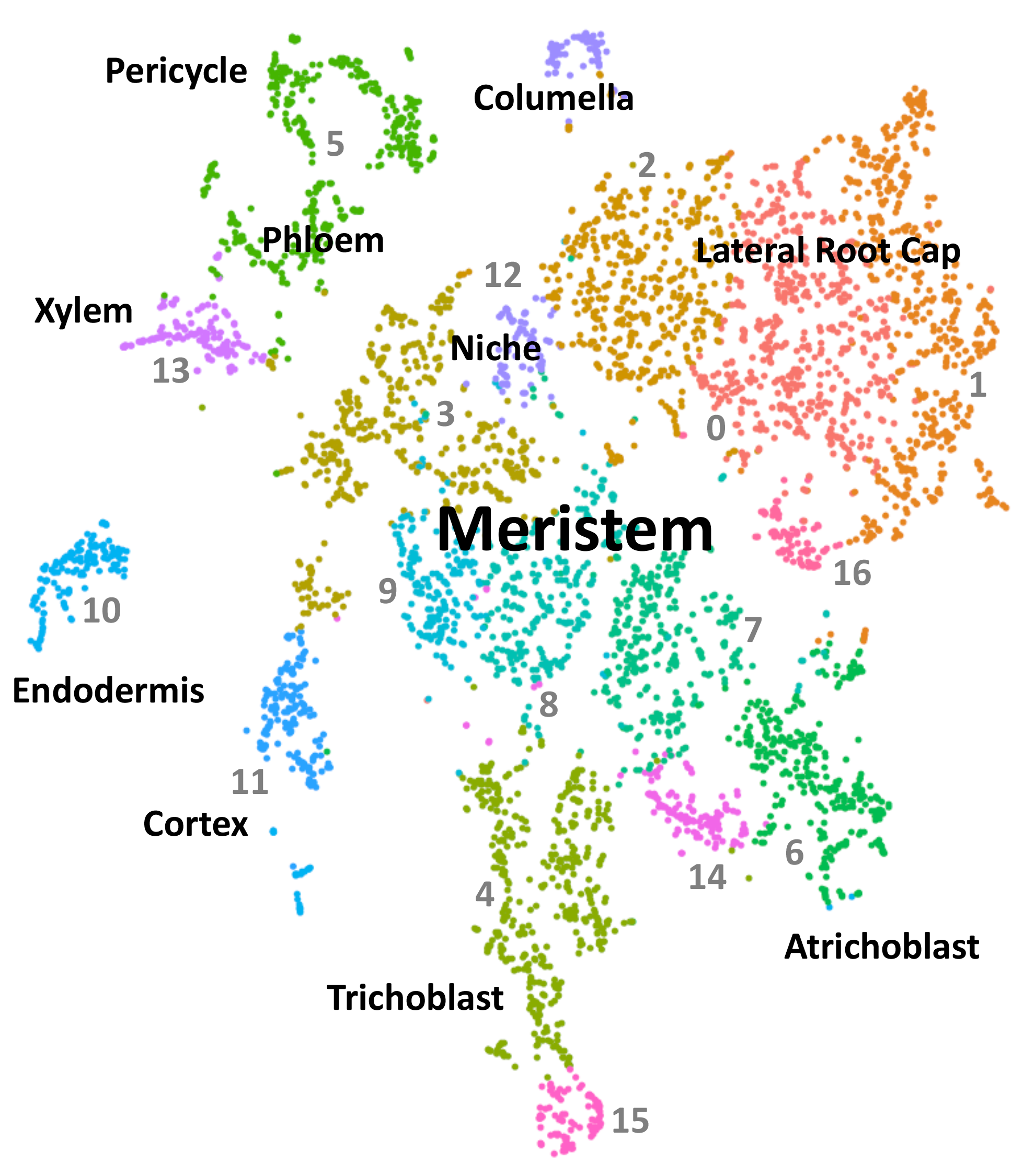
Now we have the data in tidy format, with only the average expression values. But, in order to map them into a ggPlantmap, we need to refer each cluster to a specific cell type. According to the Arabidopsis Root with protoplasting-induced genes dataset, each cluster is annotated to a specific cell-type based on unique molecular markers.
reference <- read.table("data/Summary of celltypes Arabidopsis Root.txt",fill=T,sep="\t",header=T)
reference## Cluster Identity
## 1 C0 Lateral Root Cap
## 2 C1 Lateral Root Cap
## 3 C2 Lateral Root Cap
## 4 C3 Meristem
## 5 C4 Trichoblast
## 6 C5 Phloem
## 7 C5 Pericycle
## 8 C6 Atrichoblast
## 9 C7 Meristem
## 10 C8 Meristem
## 11 C9 Meristem
## 12 C10 Endodermis
## 13 C11 Cortex
## 14 C12 Columella
## 15 C13 Xylem
## 16 C14 Unknown
## 17 C15 Trichoblast
## 18 C16 Lateral Root Cap
This reference table was generated based on the image provided in the database. We created a rough association for demonstration purposes. This is an extremely challenging part, as the distinction between clusters is not clear in many datasets.
{kind=link}
Ok, now we can proceed in plotting the expression values in a ggPlantmap heatmap. We will use the pre-loaded ggPm.At.rootmatur.crossection map
ggPm.At.rootmatur.crosssection## # A tibble: 1,113 × 5
## ROI.name ROI.id point x y
## <chr> <int> <int> <dbl> <dbl>
## 1 Atrichoblast 1 1 968. -323.
## 2 Atrichoblast 1 2 987. -305.
## 3 Atrichoblast 1 3 999. -300.
## 4 Atrichoblast 1 4 1019. -320.
## 5 Atrichoblast 1 5 1027. -332.
## 6 Atrichoblast 1 6 1024. -353.
## 7 Atrichoblast 1 7 1012. -366.
## 8 Atrichoblast 1 8 994. -369.
## 9 Atrichoblast 1 9 980. -367.
## 10 Atrichoblast 1 10 972. -365.
## # ℹ 1,103 more rows
ggPlantmap.plot(ggPm.At.rootmatur.crosssection)
The example below show a for loop that will generate a plot for every unique gene in the processed.data object
for (k in unique(data$X)) {
single.data <- processed.data %>%
filter(X == k) %>% ## filtering for one gene
merge(reference,by.x="name",by.y="Cluster",all.x=T) ## combining with the reference dataset for labeling clusters
## Merging with the ggPm.At.rootmatur.crossection using the ggPlantmap.merge() function
final.table <- ggPlantmap.merge(ggPm.At.rootmatur.crosssection,single.data,id.x = "ROI.name",id.y="Identity")
## Using the ggPlantmap.heatmap to create a heatmap
plot <- ggPlantmap.heatmap(final.table,value.quant = value) +
scale_fill_gradient(low="white",high="red",limits=c(0,1.5)) +
labs(title=paste0(k)) ## Title of the plot should be name of the gene
print(plot) ## printing the plot
##ggsave(plot=plot,paste0(k,".png"),dpi=300) ## in case you want to save your heatmap
}


